2.1 Molecular Modeling
2.1.4 Energy strain
Steric strain in protein three-dimensional structures is related to unfavorable inter-atomic interactions
and may be due to packing or functional requirements, or indicate errors in a structure's coordinates.
Detailed energy functions are usually considered too noisy for error detection. After a short energy
refinement, a full-atom detailed energy function becomes a sensitive indicator of errors. Statistics of
the energy distribution of amino acid residues in high-resolution crystal structures represented by models
with idealized covalent geometry were calculated. Interaction energy of each residue with the whole
protein structure and with the solvent was considered. Normalized deviations of amino acid residue
energies from their average values were used for detecting energy-strained and, therefore, potentially
incorrect fragments of a polypeptide chain. Protein three-dimensional structures of different origin (X-ray
crystallography, nuclear magnetic resonance spectroscopy, theoretical models and deliberately
misfolded decoys) were compared. Examples of the applications to loop and homology modeling are
given. Elevated level of energy strain may point at a problematic fragment in a protein
three-dimensional structure of either experimental or theoretical origin. The approach may be useful in
model building and refinement, modeling by homology, protein design, folding calculations, and
protein structure analysis.
[Folding & Design - 1998]
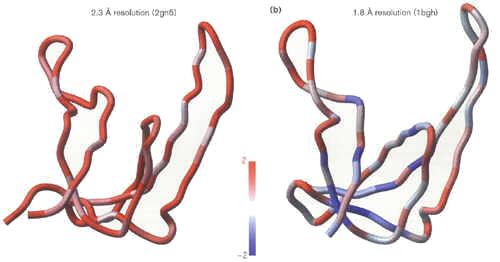 |
| Two structures of gene V binding protein solved in the same crystal form.
Left: PDB code 2gn5, resolution 2.3Ĺ; Right: PDB code 1bgh, resolution 1.8Ĺ. Residues are colored according to the
normalized residue energy (NRE) values. |