2.4 Methods and Algorithms
2.4.3 Modeling by homology
Five models have been built by the ICM method for the Comparative Modeling section of the Meeting
on the Critical Assessment of Techniques for Protein Structure Prediction. The targets have homologous
proteins with known three-dimensional structure with sequence identity ranging from 25 to 77%. After
alignment of the target sequence with the related three-dimensional structure, the modeling procedure
consists of two subproblems: side-chain prediction and loop prediction. The ICM method approaches
these problems with the following steps: (1) a starting model is created based on the homologous
structure with the conserved portion fixed and the nonconserved portion having standard covalent
geometry and free torsion angles; (2) the Biased Probability Monte Carlo (BPMC) procedure is applied to
search the subspaces of either all the nonconservative side-chain torsion angles or torsion angles in a
loop backbone and surrounding side chains. A special algorithm was designed to generate low-energy
loop deformations. The BPMC procedure globally optimizes the energy function consisting of ECEPP/3
and solvation energy terms. Comparison of the predictions with the NMR or crystallographic solutions
reveals a high proportion of correctly predicted side chains. The loops were not correctly predicted
because imprinted distortions of the backbone increased the energy of the near-native conformation
and thus made the solution unrecognizable. Interestingly, the energy terms were found to be reliable
and the sampling of conformational space sufficient. The implications of this finding for the strategies of
future comparative modeling are discussed.
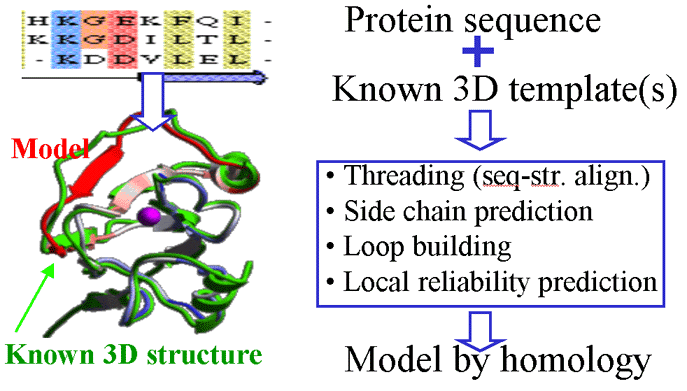 |
| General scheme of the modeling by homology method |
[Proteins - 1995]