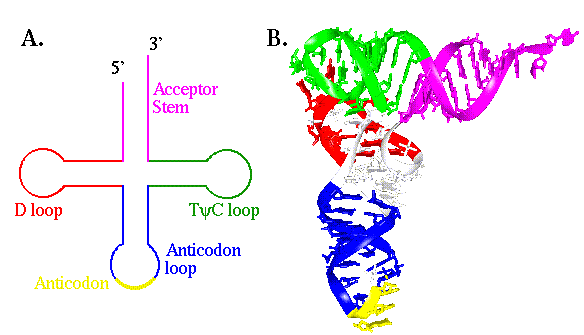
Transfer RNA molecules (tRNAs) are typically about 75 nucleotides long and fold into stable tertiary structures not unlike polypeptides (Figure 8.1). While they have no independent chemical function, their structures are finely tuned to suit a number of steps in translation. They must be specifically recognized by the enzymes that attach amino acids to them (see Exercise Ten), they must bind efficiently to the catalytic sites in the ribosome and they must form productive interactions with mRNA transcripts.
Figure 8.1 Two representations of the structure of tRNA. (A) A cartoon showing the cloverleaf-like secondary structure of tRNA. They are composed of 4 stems and three loops. (B) A three dimensional model of tRNA(Phe) from yeast, showing how the secondary structure folds into tertiary structure. The color coding is the same as for "A".
The focus of this exercise will be on the adaptation of tRNA structure that permits efficient and specific interactions with mRNA. The region of the tRNA molecule that interacts with the messenger is known as the anticodon. It is comprised of three nucleotides that form part of the anticodon loop. These three bases are free to hydrogen bond with other nucleotides, particularly a set of three base sequences (codons) on mRNA that correspond to the amino acid for which the tRNA molecule is specific. In principle, all three bases of the anticodon could interact via normal Watson- Crick base-pairing to form a three base pair duplex with a single codon sequence. In fact, the first nucleotide of the anticodon, position 34 in tRNA(Phe), which pairs with the third base of the codon, is known as the "wobble" base. The degeneracy of the genetic code means that several codons can specify a single amino acid. To reduce the number of tRNAs necessary to read the codons, the wobble position is often intended to base pair with two or three different bases at the 3' end of the codon. This reduces the specificity of interaction between the tRNA molecule and the transcript, and has the potential to lead to significant errors in translation if only two base pairs provide the thermodynamic stability to determine the interaction.
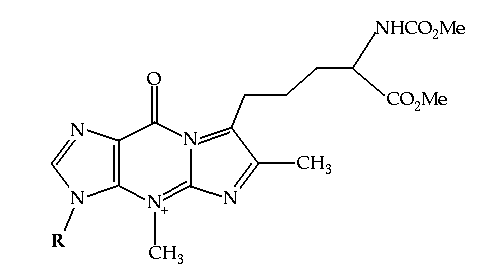
Figure 8.2 The structure of Wybutosine, a highly modified guanine analog that appears at position 37, 3' to the anticodon, in yeast tRNA(Phe).
It appears that one approach taken by nature to enhance the specific base pairing interactions between tRNA and mRNA is to use modified bases at positions flanking the anticodon in the tRNA molecule. tRNA is notable for the number and variety of modified bases in its structure. These bases are typically modified forms of the usual A, U, C and G's. Some are methylated analogues (for example 5-methyl uracil - otherwise known as thymine - is found in tRNA), and others are more dramatic, such as wybutosine (Figure 8.2). These modifications are post-transcriptional. They are made after the tRNA molecule is transcribed from its gene. The purpose of this exercise is to investigate how modifications made to tRNA(Phe) from yeast, including the creation of wybutosine adjacent to the anticodon, aid in the function of that molecule.
tRNA(Phe) has an anticodon sequence GAA, that binds to sequences UUC and UUU on the messenger (both codons for phenylalanine). Because of the wobble at position 3 of the codon, and the U:A base pairs at positions 1 and 2, the interaction between tRNA(Phe) and mRNA is weaker than most. Here, we will examine how this potential problem is overcome.
Three models are required for this exercise, though a number of others will need to be created in the course of the work. The first comes from coordinates for tRNA(Phe) deposited to the PDB by Kim and co-workers.(1) 6tna.pdb is a slightly modified version of the actual PDB entry, with a few changes made for ease of manipulation in Midas. The The second model (arna.pdb is a undecamer duplex of RNA, derived from idealized coordinates of RNA in the A conformation, containing the sequence UUC at the 5' end of one of the strands. brna.pdb is a decamer of duplex RNA, like rna.pdb, but in the B conformation.
In order to see how wybutosine might stabilize the complex between tRNA(Phe) and the codon UUC on an mRNA transcript, the first thing to do is to align the the codon and the anticodon. This can be done by hand (a tedious method with some inherent inconsistency) or using an appropriate Midas command, match.
Open 6tna.pdb as model 0 and arna.pdb as model 1. Since the duplex has the sequence UUC at the 5' end of strand A, the 3' end of strand B will have the same sequence as the anticodon (GAA). The anticodon in tRNA(Phe) spans residues 34-36. The "anticodon" sequence in the RNA model spans nucleotides 9b-11b. To perform a least squares fit to superimpose the two models, type:
Return to the comparison of rna.pdb and 6tna.pdb before proceeding.
Having superimposed the duplex rna model with tRNA(Phe), one can now begin to examine the role of wybutosine. As a first step, the conformation of the long exocyclic sidechain (atoms C13-C24) substituted at C12 needs to be adjusted, now that the complementary strand has been added. It may be helpful to reduce the number of displayed atoms to only nucleotides 34-37 from model #0 and nucleotides 1a-3a from model #1. Using the rotation command on the appropriate dihedral bond angles, adjust the sidechain to accomodate the duplex. (Note that the dihedral between N20 and C21 should be constrained to 0 or 180 degrees). Are any positive, stabilizing interactions available between the sidechain and the "codon"? When the conformation has been adjusted, fix all rotations by typing:
Where X is the rotation number. Then save the two models in their current orientations. This is done using the write command:
Where the first digit is the model number. The entry "relative 0" indicates that the coordinates for the model should be written in model 0's frame of reference. The new filename is given at the end of the command.
Finally, determine how effective wybutosine is at stabilizing the first base pair of the codon-anticodon duplex via base stacking. This can be done by measuring the surface area of the base pair that is buried by wybutosine, using dms. A single file must be created that contains both the tRNA(Phe) molecule and the "codon". Using a text editor (jot is convenient on an SGI), create a combined file by cutting and pasting the coordinates for nucleotides 1a-3a from rna2.pdb into 6tna2.pdb file. Also edit out the lines for base 37 (wybutosine) corresponding to the side chain (C14-C24, C13 can remain because it lies in the plane of the base), since these are not involved in base stacking. Save this combined set of coordinates as duplex.pdb.
Before running dms, create an input file (bp.fil) that specifies the nucleotides involved in the base pair that stacks with wybutosine. It need only specify two bases, as follows.
(Make sure all base atoms are included.) Then invoke dms
The file wybut.log will contain the solvent accessible surface area of the two nucleotides. Now compare this value to the surface area that would be accessible if only a simple, underivatized purine were used, such as adenine. Open duplex.pdb in Midas and mutate base 37 to adenine, using the swapna command:
Note that swapna is directly analogous to the swapaa command. Then use the write command as above to save the new duplex file as duplexA.pdb. Use dms a second time to calculate the accessible surface area with adenosine in place of wybutosine:
What is the percent increase in surface area that becomes exposed if a standard purine is left at position 37? To see the surfaces that have been calculated, open Midas and type:
Initially, only the model will appear, but to see the surface as well, type:
1. S. R. Holbrook,J. L.Sussman, R. W. Warrant, S.-H. Kim (1978) J. Mol. Biol. 123, 631