| 2 Research | |
| Section Intro | flexible protein-ligand docking | Virtual screening | flexible protein-protein docking |
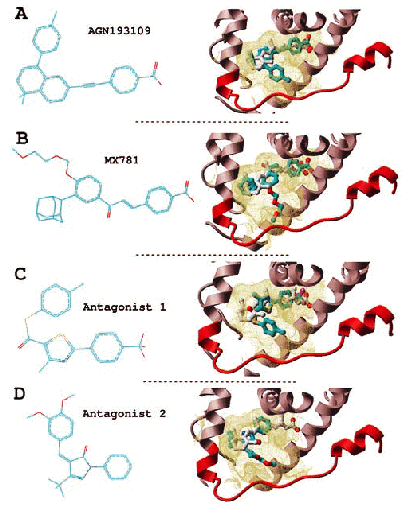 |
| RAR antagonists. Two known antagonists (A and B) and two novel antagonists (C and D). (Left) Chemical structure. (Right) Conformation docked into the receptor (part of the receptor is displayed as a ribbon representation, and the binding pocket boundary is displayed in yellow). Cyan, carbons; red, oxygen: blue, nitrogen; magenta, flourine; yellow, sulfur. Hydrogens are not represented for clarity. |
One of the main approaches that computational biology has pursued with high success is the simulation and prediction of flexible protein-ligand and protein-protein docking.
This technology has been achieved by combining an enourmous amount of state-of-the-art scientific data of structural biology, chemistry and statistics into computational algorithms.
Flexible protein-ligand docking are extremely important for the discovery of new drugs. Through the exhaustive integration and fine tuning of different variables, the latest programs and algorithms (such as ICM, for instance) are capable of predicting the behavior of chemical compounds and protein molecules in order to better help researchers find a more efficient drug leads. This method significantly reduces the necessary cost money and time consummed, as well as minimizing the non-specific interaction of drug molecules (and thus reflecting in reduced side-effects)
Protein-protein interactions are also extremely important, since they are responsible for many necessary biological functions. Prediction of such interactions is extremely important to the complete understanding of human physiology.