| References | Versions | Obtaining | Movies | Documentation | Examples | Parameters | FAQ | Useful Tools | Work in Progress |
Much Faster Docking
Recent improvements in the code and schedule parameters mean that we can now perform 10 LGA dockings in about 15 minutes, (using a Silicon Graphics 250MHz Octane). Before, they used to take around 7 hours (using a Silicon Graphics 250MHz Reality Engine). The success rate is comparable: 9 out of 10 dockings match the crystal structure, in a test with XK-263, a cyclic urea HIV-1 protease inhibitor docking to the HIV-1 protease in 1HVR. This opens up the door to the possibility of screening libraries.Adaptive Search Technques
The new optimization methods come out of an on-going collaboration with Professor Rik Belew from the Department of Computer Science at UCSD. The hybrid GA is based on the doctoral work of William E. Hart, on ``Adaptive Global Optimization with Local Search''.Incorporation of desolvation free energy
This has been achieved using atomic solvation parameters using the method of Stouten et al., and is now built into AutoGrid 3.0.Empirical free energy function
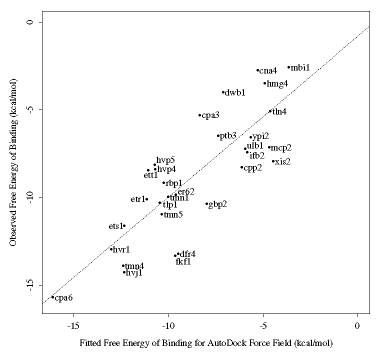
We have carefully investigated a variety of possible terms for predicting free energy of binding, using S-PLUS, a statistical analysis package. The model was callibrated on a set of diverse, structurally known ligand-protein complexes. The best model, out of a total of 900, was able to predict binding free energies to within 2 kcal/mol. This is sufficient to distinguish ligands with Ki's that are millimolar, micromolar or nanomolar: particularly important in drug design. A graph showing the correlation between observed and predicted free energies of binding is shown below. The Brookhaven PDB accession codes, in the form of three letters followed by the number, are shown also.
The movies described here
were created from work done with AutoDock 2.4. This work has been
published in the Journal of Computer-Aided Molecular Design, see
the References
page. You will also find several publications from some of the groups that
have obtained AutoDock, showing diverse applications of AutoDock.
AutoDock 2.2 was released until 21 October, 1996. It is still
available if you need it, but version 2.4 has more features and is more
robust. We recommend obtaining 2.4.
![]()
| References | Versions | Obtaining | Movies | Documentation | Examples | Parameters | FAQ | Useful Tools | Work in Progress |