|
Any molecular dynamics simulation that runs in NAMD can be used for
Interactive Molecular Dynamics (IMD). This tutorial describes
exactly what you need to do to make make a normal simulation
interactive. If you do not currently have a NAMD simulation, but you
still want to try out IMD, we provide an example simulation for you to
try below.
Step 1: Obtain NAMD
Obtain a recent copy of VMD (version 1.4b1 or
higher), and a copy of NAMD (version 2.1b1
or higher).
Step 2: Set up your simulation Prepare your simulation for
use with NAMD. If you would like to use our example simulation
instead of one of your own, download either alanin.zip or alanin.tgz. Unpack it and use the
files according to the instructions below.
Step 3: Modify your NAMD configuration file IMD
Add the following lines to your NAMD configuration file
(for example, alanin.conf):
IMDon yes
IMDport 2030
IMDfreq 1
IMDwait on
The NAMD manual
contains more information on the meaning of these parameters. Once
your configuration file is ready, run NAMD.
namd alanin.conf
NAMD will initialize your system then display the message
Info: INTERACTIVE MD AWAITING CONNECTION
indicating that it is waiting for VMD to connect.
Step 4: Load your system in VMD
Start VMD and load a PDB file (or, if you prefer, both the PDB and
PSF files) for the same system that is currently running in NAMD.
Step 5: Connect to NAMD
Open the tracker form, and enter the name of the computer running NAMD
(node 0 for a cluster) and the port you chose in the configuration
file (2030). Click "Connect". After a few seconds, you should see
your molecules start to move: you are watching your simulation in real
time!
Step 6: Interacting with your simulation
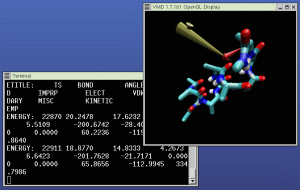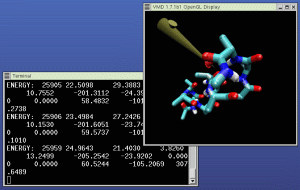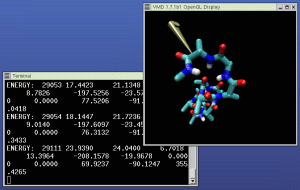 Open the mouse menu, and select "Object Mode -> Force". Select
"Atom", "Residue", or "Fragment". Your mouse can now be used to apply
forces to your simulation. Click on an atom, residue, or fragment and
drag to apply a force. Click quickly without moving the mouse to turn
the force off. You can also use a variety of 3D position trackers to
apply forces to your simulation.
Trackers with force-feedback such as the Sensable PHANTOM allow you to
feel the forces you are applying to your molecules, as if they were
real objects. See the VMD User's Guide for
information on configuring trackers.
Open the mouse menu, and select "Object Mode -> Force". Select
"Atom", "Residue", or "Fragment". Your mouse can now be used to apply
forces to your simulation. Click on an atom, residue, or fragment and
drag to apply a force. Click quickly without moving the mouse to turn
the force off. You can also use a variety of 3D position trackers to
apply forces to your simulation.
Trackers with force-feedback such as the Sensable PHANTOM allow you to
feel the forces you are applying to your molecules, as if they were
real objects. See the VMD User's Guide for
information on configuring trackers.
The picture at right shows NAMD (in the left window) and VMD (in
the right window) running our example IMD simulation. A 3D input
device is being used to pull the molecule in the simulation around.
Step 7: Disconnecting from NAMD When you are finished
interacting with your simulation, open the tracker form in VMD and
click "Detach". Your simulation will wait for another VMD
connection. If you would like to stop the simulation instead, use
"Kill".
Step 8: Further information For more information about IMD, see
the main IMD
website or the VMD
homepage. Please send questions about IMD to vmd@ks.uiuc.edu.
|