Exercise Five explores the interactions between a protein and a small molecule ligand in order to demonstrate how binding specificity is determined. Both shape and charge complementarity are examined.
This exercise will extend the use of molecular surfaces to include an electrostatic potential mapped on to the solvent accessible surface of a ligand binding region.
Molecular recognition is one a central area of inquiry in structural biochemistry. Specific interactions between proteins and their ligands are critical to biological function. Virtually every globular protein functions, in whole or in part, through specific interactions with other protein and non-protein molecules in the cell. One of the essential keys to understanding structural biochemistry is to see the patterns associated with protein-ligand interactions.
Recent developments in pharmaceutical chemistry have capitalized upon a growing understanding of such interactions to develop drugs that specifically bind to and block the activity of certain proteins. Drugs that act in such a fashion are not new, and in fact they represent a majority of pharmaceutical agents. For example, AZT binds to and blocks the function of the protein in HIV responsible for replicating the viral chromosome. In developing these drugs, however, chance played a larger role than target-specific design. With the rapid increase in structural information pertaining to proteins involved in a variety of disease-related processes, however, it is now possible to expect to attack such problems through rational methods.
One recent success in such an effort was reported by a large Australian group, lead by crystallographer Peter Colman and medicinal chemist Mark von Itzstein, working to produce a specific agent against the influenza virus.(1,2) One of the virus's coat proteins, sialidase (also referred to as neuraminidase), is responsible for hydrolyzing sialic acid from oligosaccharides (short chains of carbohydrate subunits) on the surface of cells. This activity is believed to assist the virus in penetrating the mucosal lining of the respiratory tract, and thereby infecting the host. The problem with most drugs against the influenza virus is that their specific binding sites are constantly evolving in the viral population, so that in any given year, the virus may escape effective treatment by simply losing the drug binding site. In the approach reported by von Itzstein et al. the drug is designed to specifically interact with a portion of the protein that is essential for viral replication, and therefore cannot mutate without damaging the viability of the virus. Previous attempts to develop anti-influenza drugs along these lines have suffered because of poor dissociation constants, weaker than 1e-6 M.(3)
This most recent study in rational drug design was founded upon a crystallographic study performed on sialidase complexed with sialic acid and 2-deoxy-2,3-didehydro-D-N-acetylneuraminic acid (DAN), an inhibitor developed in 1969, shown in Figure 5.1.(4) Based on their inspection of the ligand binding site in sialidase isolated from influenza virus, Colman's group decided to replace the hydroxyl group at C4 of DAN with the guanidinium group, which is bulkier and carries a positive charge. Their insight resulted in a decreased dissociation constant of about 1e-10 M (as opposed to ca. 1e-5 M for DAN). More importantly, 4-guanidino-DAN (4GDN) is not tightly bound by mammalian sialidases, where the dissociation constant is only 1e-5 M. DAN is bound equally well by both viral and mammalian proteins. That means that 4GDN can act specifically against the viral invader, without interfering significantly with the host's sialidase. In this excercise you will inspect the structure of the complex of DAN bound to sialidase and analyze the interactions present between ligand and protein. By investigating these interactions, it should be possible to see the logic behind the development of 4GDN as a specific anti-influenza agent.
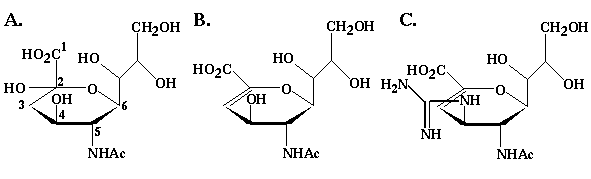
Figure 5.1 The structures of three ligands with high affinity for sialidase. (A) Sialic acid is a natural ligand for sialidase. It is a nine carbon carbohydrate that forms glycosidic linkages through the hydroxyl group on C2. (B) 2-deoxy-2,3-didehydro-D-N-acetylneuraminic acid (DAN) is an analog of sialic acid that readily adopts a ring conformation conducive to strong interactions with sialidase. (C) 4GDN is the molecule designed by von Itzstein et al. Note that at pH 7, the carboxylic and guanidine groups would normally be ionized.
Three model files are useful for this exercise: sialidase.pdb, DAN.pdb and GDN.pdb. The former file contains the coordinates deposited by Bossart-Whitaker et al.(5) and shows the complex of sialidase with DAN, derived from crystallographic data collected to 2.8 Å.(6) The model in sialidase.pdb has been altered, in that all hydrogen atoms have been removed from the structure and the bound inhibitor, DAN, has been editted out and placed in a separate pdb file... DAN.pdb and GDN.pdb are models of DAN and 4GDN (the latter model is based on the former and is not from experimental data).
Open sialidase.pdb and DAN.pdb simultaneously in Midas. Compile a table of the interactions between DAN and sialidase. It is easier to view only a subset of the entire protein by focusing on a limited region. To do so, type:
and only those residues within six Å of the inhibitor will be displayed.
It may be helpful to list the interactions in a table. List the atoms in DAN in the first column and then list the contacted residue atoms of sialidase in the second column, with distance in the third column and the type of interaction in the fourth column:
DAN Sialidase Distance Interaction O9 Glu278@OE2 3.0 Å Ion-dipole
Be careful not to overlook the possibility of van der Waals contacts. When this is complete, Midas can be quit while the next steps are completed.
While identifying individual interactions is an important step in determining specificity, it is sometimes useful to look at the overall electrostatic conditions in the binding pocket to predict ways of increasing specificity for an inhibitor. This is possible using the program esp in conjunction with dms. Together these programs can be used to create a color-coded map of the active site. Since the region of interest is limited to the DAN binding pocket, it isn't necessary to calculate the surface of each residue. Instead, a file containing a list of all residues within 6 Å of DAN (6A.fil) has been prepared as an input file for dms. (Or alternatively, this list could be prepared independently). Save this file to the working directory and in an open shell, type:
(The program will take a few minutes to run.) The "-n" flag instructus dms to prepare data that can be read by esp. Note that, because DAN is not present in the structure file (sialidase.pdb), the binding pocket can be mapped by the probe. After dms has calculated the surface, then run esp.
When this has completed move on to the next step.
To view the surface, start Midas and once it is running, type:
This opens the model of sialidase linked to the calculated surface. Then open the model of DAN, so that it is displayed in the active site. Finally open the surface, by typing:
The model can be colored and edited as usual to focus on the region of interest. When that's been done, there are several options for coloring the surface. Two are listed here:
This will first color the surface white, and then color only the most negative regions red. Alternatively, one can type
Which will color the surface in a continual fashion from red at the lowest potentials to blue at the highest.
Having colored the surface, identify whether or not it seems likely that the addition of a positively charged functional group at C4 would enhance binding of DAN to sialidase. As confirming evidence, open GDN.pdb as a third model and determine how well the guanidium group fits into the binding pocket. The guanidinium group may be rotated to enhance its fit.
2. von Itzstein, M, Dyason, J C, Oliver, S W, White, H F, Wu, W-Y, Kok, G B and Pegg, M S (1996). A study of the active site of influenza virus sialidase: an approach to the rational design of novel anti-influenza drugs, J. Med. Chem., 39, 388-391.