Exercise Four introduces tertiary structure, and specifically examines the role of disulfide bridges and the hydrophobic core in stabilizing the folded conformation of BPTI.
This exercise introduces the chain command, as well as the programs conic and dms, which are useful in inspecting exposed regions of a folded protein.
Bovine pancreatic trypsin inhibitor (BPTI) has frequently been referred to as the "hydrogen atom" of biochemistry. Just as the simplicity of hydrogen permits exact quantum mechanical solutions, structural biochemists have hoped that the small size of BPTI will lead to a clear cut understanding of the stability of protein conformation, at least in one case. This exercise is intended to explore those aspects of BPTI's structure that lead to its structural stability.
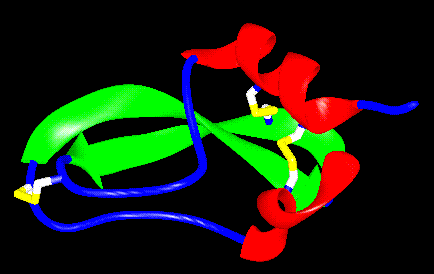
Figure 4.1 Ribbon drawing of BPTI. The disulfide bridges are shown in yellow, the beta strands in green and the alpha helices in red.
BPTI is a 58 amino acid polypeptide that adopts a tertiary fold comprising two strands of antiparallel beta sheet and two short segments of alpha helix (Figure 4.1). In addition, three disulfide bonds are formed in the stable fold, linking cysteines 5 & 55, 14 & 38 and 30 & 51.
As with any protein, BPTI's tertiary structure is, in part, stabilized by a hydrophobic core of amino acid residues that become protected from solvent after the peptide chain collapses into its folded conformation. However, BPTI's unusually small size requires that additional stabilizing features be present, since only a small number of hydrophobic residues can be effectively buried in such a small protein. Hence the presence of the disulfide linkages, which link residues widely separated in sequence close to one another in three dimensional space. Cystine linkages are commonly found in small, monomeric, extracellular proteins, where tertiary interactions are insufficient, and quaternary interactions non-existent.
To better identify those structural elements that are absolutely essential for maintaining BPTI's fold, Peter Kim and Terry Oas synthesized a synthetic peptide model of BPTI, designed only to include the C-terminal a helix and the two strands of beta sheet.(1) This model, the dipeptide PaPb (Pa is a peptide that includes residues 43-58, Pb is a second peptide comprising residues 20-33) also contains the [30-51] disulfide bridge. From previous studies, it has been shown that the [30-51] disulfide is the first to form in the folding process. Using CD studies and 1H NMR, Oas and Kim were able to show that their peptide model did in fact adopt secondary structure in solution, so long as the [30-51] disulfide bond was present. In the absence of that linkage, after the disulfide had been reduced, no secondary structure was apparent. Clearly, a large fraction of the necessary interactions to stabilize BPTI are present in PaPb, making these portions of the protein sequence a good place to begin to look for core residues.
To determine whether a residue can properly be assigned as part of the hydrophobic core, it is important to be able to evaluate its solvent exposed surface area in the context of the protein, relative to the surface area of the isolated residue (Table 4.1). If a residue is truly stabilizing a protein through the hydrophobic effect by being hidden from solvent, then presumably it has very little of its surface area exposed. As a rule of thumb, any hydrophobic residue that is 70% buried may be thought of as a core residue. To make quantitative determinations of buried surface area, this exercise will make use of an auxiliary program to Midas, called "dms". It performs this calculation and provides visual output for inspection. In calculating solvernt exposed surface area, dms takes a sphere 1.4 Å in radius as a model for a water molecule and roles it over the surface of a residue/protein. Because of the size of this spherical probe, it is unable to fit into narrow crevices and will therefore define surfaces that are smoothed over in comparison to a typical space filling model of a molecule. That portion of the surface which corresponds to the probe contacting the residue of interest is the contacted surface. Reentrant portions of the surface are those parts that result from the probe being contacted by two or more residues or atom groups including one of interest (these are the smoothed over portions of the surface).
Table 4.1 Surface areas of the sidechains of the 20 amino acids. Note that the overall surface area is the sum of the contacted surface area and the reentrant area.
| Amino Acid | Tot. SA (sq. Å) | Cont. SA (sq. Å) | Reent. SA (sq. Å) |
| Alanine | 26.58 | 23.38 | 3.20 |
| Arginine | 111.28 | 60.99 | 50.29 |
| Aspartic Acid | 65.14 | 35.48 | 29.65 |
| Asparagine | 63.15 | 34.43 | 28.72 |
| Cysteine | 56.33 | 35.61 | 20.72 |
| ystine | 69.13 | 37.71 | 31.42 |
| Glutamic Acid | 80.14 | 43.95 | 36.19 |
| Glutamine | 78.20 | 43.01 | 35.19 |
| Glycine | no sidechain | ||
| Histidine | 92.11 | 54.74 | 37.37 |
| Isoleucine | 84.77 | 51.70 | 33.06 |
| Leucine | 86.69 | 53.09 | 33.60 |
| Lysine | 95.47 | 55.07 | 40.48 |
| Methionine | 91.57 | 56.22 | 35.35 |
| Phenylalanine | 110.14 | 65.97 | 44.17 |
| Proline | 61.10 | 44.77 | 16.33 |
| Serine | 34.02 | 24.78 | 6.17 |
| Threonine | 50.39 | 35.19 | 15.20 |
| Tryptophan | 139.36 | 80.55 | 58.80 |
| Tyrosine | 116.81 | 67.54 | 48.47 |
| Valine | 65.06 | 44.18 | 20.88 |
The file bpti.pdb contains the coordinates for the crystal structure of BPTI solved by Huber and coworkers.(2) Solved to 1.5 Å resolution, with an R-factor of 16.2%, this is one of the more reliable sets of atomic coordinates available for a protein. Solvent molecules that cocrystallized with the protein have been stripped from this model for the sake of simplicity.
dms is a program that runs outside of Midas but produces files that can be visualized in Midas. It is fully described in the Midas manual on pages 103-4. To perform this experiment, the model of bpti must be saved to the working directory. In addition an input file needs to be used in order to indicate which residue(s) are to have their surface areas calculated. A sample file is available and can be saved to the working directory. Then dms can be run in the console window. To run dms, type the following information at the console prompt and hit return:
"model.pdb" is the pdb file containing the protein structure whose surface, in part, is to be calculated. "model.log" is the log file from dms that will provide the information regarding the calculated surface (-g indicates to dms that this is the log file). To read the contents of "model.log", type (in a shell window):
The contents of the log file appear something as follows:
The critical information from the log file is that the surface being calculated was 75.09 sq. Å in area, with 27.95 sq. Å of that being at points where the water would contact more than one atom or atom group. "model.fil" specifies the portion of the molecule to be used in calculating a surface (-i indicates this as an input file). In this exercise it will be the side chain atoms for a single residue. (The total surface area of the protein will be calculated if the input file is omitted.)
To generate an appropriate file to calculate the surface of the side chain of Valine 7, for example, use jot to edit the following file:
"model.dms" is called the surface file, and actually contains the information that Midas will need to allow visualization of the solvent exposed surface (-o identifies it as an output file).
As best as possible, prepare a list those hydrophobic residues that represent the "core" for BPTI. This procedure can be somewhat tricky, given the complexity of even a small protein. Since it is already known that a functional core is available in PaPb, it is useful to concentrate on those peptides. To simplify the model, first type:
which will give an alpha carbon trace of the protein backbone. Secondly, add back those residue side chains in PaPb by typing:
The disp(lay) command adds on the specified residues, whereas the "show" command erases the screen and shows only the specified residues. Color the model and assign core residues.
Confirm at least three of the core residue assignments by doing an accessible surface calculation for the side chain of a core residue using the dms program. To begin this exercise, it may be most useful to use the show command to display all the residues (since other portions of the protein help bury the core). The conic routine is also useful in determining qualitatively if a residue is buried. Conic prepares a static, space-filling image of the protein. Color the residue in question some unusual hue and then type
If that color is substantially masked by other residues, you may have found a core residue, and dms can do the rest.
To use dms, copy the model.fil file into the working directory and edit it, using jot, to include all the sidechain atoms of the residue of interest. Save the file using the necessary commands under the "File" menu. In the console window type:
Edit the bpti.log file to uncover the actual exposed surface area of the residue.
BPTI folding intermediates containing two disulfides, namely [14-38,30-51] and [14-38,5-55], possess largely native tertiary structure. However they are slow to form the third disulfide that is found in the native structure. Why might that be the case? Consider the mechanistic problems associated with disulfide formation and the structural constraints that might be placed on the remaining free cysteines. (Hint: examine the solvent accessibility of [30-51] and [5-55])
(1) T. G. Oas and P. S. Kim (1988) Nature, 336, 42-48.
(2) M. Marquat, J. Walter, J. Deisenhofer, W. Bode and R. Huber (1983) Acta Crystallographica 39, 480.