Exercise One focuses on intermolecular contacts between molecules in a crystalline lattice. Ionic, hydrogen-bonding and van der Waals contacts will be investigated.
This exercise will cover opening and closing pdb files in Midas, rotating and translating images in Midas with the mouse, coloring images and determining distances between atoms.
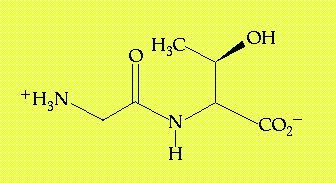
Exploring the interactions between atom groups within or between large molecules will be a central activity in these exercises. It's these interactions that provide the basics for macromolecular conformation, molecular recognition and catalysis. Despite the focus here on large biopolymers, the interactions that lead to biochemical specificity are the same for all molecules. In this exercise, we will focus on a relatively simple chemical species, the dipeptide glycyl threonine (GT, Figure 1.1), crystallized in the dihydrate form (the overall chemical composition of the crystal indicates two molecule of water crystallized with every molecule of the dipeptide.)
Figure 1.1 - The structure of glycyl-L-threonine. There are also two water molecules associated with this compound in its crystalline state.
Crystals form through the slow precipitation of a chemical species, such that each molecule adopts an orientation on the surface of the growing crystal that takes best advantage of the available intermolecular interactions. For example, in an ionic crystal, cations always locate themselves so as to be surrounded by anions. In a molecular crystal, such as formed by GT, ionic interactions are coupled with hydrogen bonding and van der Waals contacts to lead to the most stable orientation of each molecule with respect to its neighbors. Because each molecule has the same requirements for stabilizing interactions as any other molecule in a pure molecular crystal, regularity of orientation results. In fact, it's that regularity that makes a crystal a crystal. A crystal is built of smaller units, referred to as "unit cells", that can be thought of as boxes that can be stacked on each other in all three dimensions to build up a repeating 3-dimensional structure (Figure 2.2). Therefore, one only needs to inspect a single unit cell to understand the environment that every single molecule in the crystal experiences.
In the case of glycyl threonine, there are four molecules in the unit cell, each oriented slightly differently in three dimensional space with respect to the others. However, each is related to the other by a simple symmetry operation about a central axis, so in fact each molecule feels an identical set of surroundings to every other molecule in the unit cell. In this instance, a single molecule is said to reside in the asymmetric unit (Figure 1.2). The important information to be taken from this discussion is that every molecule of GT has an identical set of intermolecular contacts to every other molecule in the crystal.
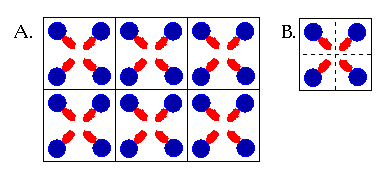
Figure 1.2 (A) A simple diagram showing how a 2-dimensional crystal can be constructed out of boxes that represent units cells. This "crystal" can be extended by adding more boxes to the sides or top and bottom. (B) A single unit cell contains four molecules, symmetrically related to one another. One quarter of each box contains the asymmetric unit of the unit cell. The asymmetric unit is that part which can be used to construct the whole of the unit cell.
Glycyl-L-threonine is of interest in this context, not only because it is a dipeptide, but also because it offers the opportunity to observe a variety of different kinds of interactions: ion-ion, ion-dipole, dipole-dipole and van der Waals. The examination of a crystal structure of a small molecule, in which the positions of the individual atoms are known with relatively high precision, provides a good opportunity to observe the positioning of various atom groups with respect to one another as specific types of interactions are formed. The geometries and distances observed here for intermolecular interactions will be repeated many times over in macromolecules.
The PDB-formatted data files to be used in this exercise are gtcell.pdb and gt.pdb. Save these files to your home directory by clicking on the links (the blue type) and choosing "Save As" from the "File" menu. The file gt.pdb contains a pdb formatted file with coordinates for 13 individual molecules of glycyl-L-threonine along with 26 co-crystallized molecules of water. These 39 molecules come from several different unit cells, but if you're interested in seeing what one unit cell looks like, that image is available in the file gtcell.pdb.
For more information on any of the italicized commands and options that are used in this exercise (and all subsequent exercises), type help "command" at the command line of Midas
Start Midas up by typing the following command in an open UNIX shell (an open command line window)
This command will open Midas with gt.pdb displayed to the screen. The atom coordinates from gt.pdb are refered to as a "model" and by default gt.pdb is opened as model #0. Other pdb files can be opened subsequently and will be assigned sequential model numbers. For example, to open gtcell.pdb once you have already opened midas with gt.pdb, type
and gtcell.pdb is opened as model #1.
These two models can be rotated and translated together or individually by using the mouse. The left mouse button controls rotation about the x (horizontal), y (vertical) and z (perpendicular to the screen) axes, while the center mouse button allows translation in the x and y plane. The images can be translated in the z axis by depressing the left and middle mouse buttons together.
Other manipulations are possible using the Midas Control Panel that accompanies the Midas window. For example, the scale of the images can be changed by clicking and dragging on the small box in the sideview window or by using the buttons associated with "scale" in the "Assignments" list (Figure 1.3). The two different models, gt.pdb and gtcell.pdb, can be manipulated either together by selecting "all" from the "select models" box or separately by selecting"individually" and then clicking on the models that are to remain unaffected by manipulations (they can be reactivated by clicking a second time.)
When you're finished viewing the cell, you may close it again by typing
By specifying the model number "1", only gtcell.pdb will be closed.
Because there are 39 different molecules stored in gt.pdb, a "chain identifier" is used (though typically chain identifiers specify different polypeptide chains). The GT molecules are labeled A, B, C and so on to M. The waters are referred to as 1A* and 2A* (* indicates that the molecule is not part of a protein or DNA molecule, it is a "heteroatom"), 1B* and 2B*, and so on. GlyThr A will be the focus of this exercise, as it is located roughly at the center of the assembly of 13 GT molecules.
Having opened the model, color the atoms by typing
This colors all the atoms in all the open models by element according to a standard color scheme:
Some additional image modification may be helpful to you in this exercise. For example, you may wish to color molecule A differently than the others. To color GlyThr A differently, try coloring the carbon atoms in that molecule a different color (red, blue, yellow, green, orange, magenta, cyan, gray, white and black are available) ie:
Note that this command defines certain atoms using Midas atom specification (atom_spec) symbols, as follows:
Note that atom names correspond to PDB conventions for amino acids and nucleotides (see Appendix B). The atom descriptor @C= uses a wildcard (=) to describe all carbon atoms with one, two and three letter names starting with "C". Another option would be to use the "?" wildcard which substitutes for a single character. For example, one could type the following with the same results as above:
The goal here is to list the two or three most significant contacts made by each atom in molecule A to other molecules in the crystal. In this table, you should have columns for
A sample entry might look like:
| 1A@N | 2J@OXT | ion-ion | 2.85 Å |
There are a number of strategies for identifying intermolecular contacts made to a particular atom. The simplest is to erase all those atoms/molecules outside a certain distance from the specified atom. For example, to identify all those atoms that might be hydrogen bonding with the nitrogen atom of glycine residue 1A, the following command may be used
It will erase all residues that lie outside a sphere of 3.5 Å. One might alternatively type
to show only the atoms that lie within that 3.5 Å sphere. The sphere size should be adapted to include those interactions of interest (see Appendix C for lists of appropriate contact distances). To refresh the screen, simply type
Measuring distances can be done using the dist(ance) command, which monitors the distances between two specified atoms. This can be used in conjunction with "picking" to speed up information gathering. At the command line, type
and then, while depressing the "Alt" key on the keyboard, click on two atoms with the left mouse button. Atom labels for those atoms will be returned automatically to the command line. Hitting return will then cause the distance to be given (along with an identifying distance number) at the top right corner of the Midas window. One can measure sixteen different distances simultaneously (numbered 0-15). To remove a distance, type
To end the modeling session, type stop at the Midas command line and log off of the computer by placing the cursor over the desktop, depressing the right mouse button and selecting "log out". The computer will query you regarding your intentions in this vein and you may respond yes.