CHARMM
The CHARMM program is a general purpose molecular mechanics and molecular dynamics simulation program. The core of the program is based on the empirical potential energy function discussed earlier which is used for energy minimization, molecular dynamics simulations, vibrational analysis and thermodynamic calculations. In addition, there are interfaces to several quantum mechanical programs which allow one to do mixed QM and MM calculations. The program contains a comprehensive analysis facility which enables the user to compare structures, evaluate energies, calculate time series and correlation functions. The program can treat systems ranging in size from small individual organic molecules to large proteins and DNA molecules in solution. It has been developed in the laboratory of Professor Martin Karplus at Harvard University over the past 20 years. Today, contributors continue to improve and enhance the capabilities of the program.
Below is a list of the main features of the latest version of the CHARMM program, earlier versions may not contain all the feature listed below.
Currently, parameter sets are available for proteins1, DNA/RNA2, and lipids3. The CHARMM program runs on many different platforms including DEC alpha, Convex, Cray, HP, IBM RISC, IBM SP, SGI, SUN, GNU and Linux machines.
A complete description of commands available in the CHARMM Program is available in the CHARMM documentation supplied with the program as well as in the CHARMM paper [J. Comp. Chem., 4, 187 (1983)]. See also http://www-esbs.u-strasbg.fr/notes-de-cours/modeli/charmm/CHARMM-docs.html for the complete documentation online. Here, we describe some of the basic data structures and introduce some of the basic commands.
|
Example of the RTF for the Alanine residue: RESI ALA0.00 GROUP ATOM NNH1-0.47 ! | ATOM HNH 0.31 ! HN-N ATOM CACT1 0.07 ! | HB1 ATOM HAHB 0.09 ! | / GROUP !HA-CA--CB-HB2 ATOM CBCT3-0.27 ! | \ ATOM HB1HA 0.09 ! | HB3 ATOM HB2HA 0.09 ! O=C ATOM HB3HA 0.09 ! | GROUP ! ATOM CC 0.51 ATOM OO-0.51 BONDCB CA N HN N CA BOND C CA C +N CA HA CB HB1 CB HB2 CB HB3 DOUBLE O C IMPR N -C CA HN C CA +N O DONOR HN N ACCEPTOR O C IC -C CA *N HN 1.3551 126.4900 180.0000 115.4200 0.9996 IC -C N CA C 1.3551 126.4900 180.0000 114.4400 1.5390 IC N CA C +N 1.4592 114.4400 180.0000 116.8400 1.3558 IC +N CA *C O 1.3558 116.8400 180.0000 122.5200 1.2297 IC CA C +N +CA 1.5390 116.8400 180.0000 126.7700 1.4613 IC N C *CA CB 1.4592 114.4400 123.2300 111.0900 1.5461 IC N C *CA HA 1.4592 114.4400 -120.4500 106.3900 1.0840 IC C CA CB HB1 1.5390 111.0900 177.2500 109.6000 1.1109 IC HB1 CA *CB HB2 1.1109 109.6000 119.1300 111.0500 1.1119 IC HB1 CA *CB HB3 1.1109 109.6000 -119.5800 111.6100 1.1114 |
The following data structures are constructed for a specific molecule or molecules.
Protein Structure File (PSF)
The Protein Structure File (PSF) is the most fundamental data structure in the CHARMM program. It is generated for a specific molecule or molecules and it is a concatenation of the information contained in the RTF file. It contains the detailed composition and connectivity of the molecule(s) of interest. It gives the total number of bonds and identifies which atoms make up a particular bond. Similarly for the bond angles, where triplets of atoms are listed, and for torsion angles, where quadruplets of atoms are specified. Improper dihedral angles are also listed; these are used to maintain planarity and prevent accidental chiral inversion. The PSF can include several molecular entities, defined as segments, which can range from a single macromolecular chain to multiple chains solvated by explicit water molecules.
The PSF must be specified before any calculations can be performed on the molecule. The PSF constitutes the molecular topology but does not contain information regarding the bond lengths, angles, etc., so it is necessary to read in the parameter file to add the missing information.
Coordinate File (CRD)
The coordinate file contains the Cartesian coordinates of all atoms in the system. The cartesian coordinates of a molecule are most often obtained from x-ray crystal structures or from NMR structures. Alternatively, they can be generated from homology modeling studies or from the library values contained in the topology and parameter files along with some secondary structure information. Missing coordinates can be built within the CHARMM program using the internal coordinate facility (see below); in addition, hydrogen atoms, which are not present in x-ray crystal structures of proteins may be placed using the HBUILD module in CHARMM. Two sets of coordinates can be accommodated in the CHARMM program, which is useful for a variety of calculations. For example, it can be used to the calculate the root mean square difference (rmsd) between a structure from the simulation and the experimental structure.
Internal Coordinate Table (IC Table)
The Internal Coordinate Table contains information on the positions of the atoms relative to one another within the structure, i.e., the bond lengths, angles and dihedrals etc. for the molecule. The IC table is useful for analyzing the structure of a molecule or for building parts of the molecule that are missing. This table can be constructed by including the setup keyword in the generate statement when generating the PSF file. The table itself has a particular notation, see below.
|
|
|
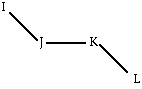 |
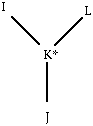 |
Each entry in the IC table (see below) lists 4 connected atoms, I, J, K and L,; for a normal IC table entry, the I-J bond length, R(IJ); the I-J-K bond angle, T(IJK); the dihedral angle I-J-K-L, PHI; the bond angle T(JKL); and the K-L bond length, R(KL) are listed. Improper dihedral angles, which are used to keep sp2 atoms planar and sp3 atoms in a tetrahedral geometry, are marked with a star. The center atom of an improper dihedral angle is marked with a star. For an improper dihedral angle entry, the I-K bond length, R(IK); the I-K-J bond angle, T(IKJ); the dihedral angle I-J-K-L , PHI; the J-K-L bond angle T(JKL); and the K-L bond length, R(KL) are listed. The atom entry "-99" indicates an undefined atom.
Example of an Internal Coordinate (IC) Table
|
N
|
|
|
|
|
|
|
|
|
|
|
1
|
|
|
|
|
|
|
|
|
|
|
2
|
|
|
|
|
|
|
|
|
|
|
3
|
|
|
|
|
|
|
|
|
|
|
4
|
|
|
|
|
|
|
|
|
|
|
5
|
|
|
|
|
|
|
|
|
|
|
6
|
|
|
|
|
|
|
|
|
|
|
7
|
|
|
|
|
|
|
|
|
|
|
8
|
|
|
|
|
|
|
|
|
|
|
9
|
|
|
|
|
|
|
|
|
|
|
10
|
|
|
|
|
|
|
|
|
|
|
11
|
|
|
|
|
|
|
|
|
|
|
12
|
|
|
|
|
|
|
|
|
|
|
13
|
|
|
|
|
|
|
|
|
|
|
14
|
|
|
|
|
|
|
|
|
|
|
15
|
|
|
|
|
|
|
|
|
|
|
16
|
|
|
|
|
|
|
|
|
|
|
17
|
|
|
|
|
|
|
|
|
|
|
18
|
|
|
|
|
|
|
|
|
|
|
19
|
|
|
|
|
|
|
|
|
|
|
20
|
|
|
|
|
|
|
|
|
|
|
21
|
|
|
|
|
|
|
|
|
|
The CHARMM input file
Although the CHARMM program can be run interactively (see below) it is often more convenient to put the CHARMM commands in a file and submit the CHARMM job in batch mode; this makes debugging the commands easier.
All CHARMM input files begin with several lines of comments. These title lines begin with a "*" as the first character and the title is terminated by a line containing only a "*". For example, in our first CHARMM input file, example1.inp, we begin as follows:
* Example 1: First CHARMM input file
* Defines and builds a PSF file, then calculates the energy
*
There are several rules for specifying CHARMM commands
CHARMM commands:
The first word in every command line specifies the command. The command line is written in free format and is generally written to the standard output. Most commands have several options, these follow the command word in the form of specific keywords. In most cases only the first four letters of the keyword are required. For clarity we will capitalize the first four characters of each command.
Example: To open and read in the topology (top_all22_prot.inp) and the parameter files (par_all22_prot.inp), the CHARMM commands are
OPEN READ FORMatted UNIT 27 NAME top_all22_prot.inp
READ RTF CARD UNIT 27
OPEN READ FORMatted UNIT 28 NAME par_all22_prot.inp
READ PARA CARD UNIT 28
The next step to setting up a CHARMM calculation is the generation of the PSF file for your particular system. First, the sequence must be specified, this can either be done by typing in the sequence or by reading it in from the coordinate file. Once the sequence has been specified, the PSF is built using the GENErate command. For example,
! Read Sequence for Polyalanine ! This is a comment line
READ SEQU CARD ! The read sequence command
* Poly-alanine sequence ! Title line
* ! End of title line
6 ! Number of residues
ALA ALA ALA ALA ALA ALA ! Specify the residues
GENErate ALA6 SETUp ! Command to generate the psf
In the example above, ALA6 is the name associated with the sequence of six alanine residues and the SETUp keyword specifies that the internal coordinate table should be constructed. By default, N and C terminal groups are added during the generate statement. We will look at these and other commands in the exercise section.
Stream files
It is often convenient in CHARMM input files to include portions of the main command file in CHARMM stream files, and call them when necessary. This helps make the input files clear and straightforward to understand. The command to read in a stream file is STREam. A stream files starts with a title and can include as many CHARMM command lines as needed. It ends with the command RETUrn.
For example, in your master command file, the following command would be included if you want read in a stream file which contains the commands do an energy minimization
STREam mini.str
In your local directory, you would have a file called mini.str which would include the following commands,
* Energy minimization protocol
*
MINI SD NSTEP 200 NPRInt 10
MINI ABNR NSTEP 500 NPRINT 50
RETUrn
Comments
Comments can be entered on new lines anywhere in the command file or at the end of any command line. A comment is indicated by an exclamation mark (!) at its beginning.
Running the CHARMM program
Interactive mode
The CHARMM program may be run interactively by first launching the CHARMM program and then entering the command lines one by one, pressing the <RETURN> key at the end of each line. CHARMM has extensive error checking, so if a command is mistyped, an error message is be provided. Often, if the error is serious enough, the CHARMM session will be terminated. You will then have to run CHARMM again and retype everything up to that final mistake. This can be a very inefficient way to run CHARMM, so it is reccommended that input files are written and submitted to CHARMM in batch mode. To terminate an interactive session, hold the CTRL key down and type c. This action will terminate CHARMM. To run CHARMM interactively simply type
charmm
The full path should be typed if the CHARMM executable is not in your local directory. You will then get a message indicating the version of CHARMM you are running, the current time and date and your name. You then type in the title lines, terminated with a lone *; you can then continue typing in different commands. The output will be directed to the screen.
Batch mode
The alternative to running in interactive mode, is batch mode. In this case, all the CHARMM commands are put in a file, for instance example1.inp. and this file is directed into CHARMM by typing
charmm < example1.inp > example1.out
The output which previously came to your screen is now directed to the file example1.out. The results can be viewed by editing this output file using an editor of your choice.