Virtual ligand screening (VLS) based on high-throughput flexible docking is an emerging technology for rational lead discovery based on receptor structure. Rapid accumulation of high-resolution three-dimensional structures, further accelerated by the structural proteomics initiative and the improvements of docking and scoring technology, are making VLS an attractive alternative to the traditional methods of lead discovery. VLS can sample a virtually infinite chemical diversity of drug-like molecules without synthesizing and experimentally testing every screened molecule. Typically, a corporate high-throughput screening (HTS)-ready compound library ranges from 200,000 to 1,000,000 molecules. Even with corporate libraries as large as these, however, the experimental HTS often does not result in viable leads (Martin Rosenberg, personal communication). The high cost of such massive experimental testing and its technical complexity are further motivation for the theoretical alternative. Finally, the virtual experiment, as opposed to a high-throughput assay, can be easily designed to select for a particular binding site or receptor specificity.
Docking and screening methods have a long history that is described elsewhere.We have not tried to cover docking and binding energy-prediction methods that are too computationally expensive for high-throughput applications and take more than 3 5 minutes per ligand per processor. Here we will review the most recent advances in the area of high-throughput flexible docking and computer screening,as well as applications of these techniques to lead discovery.
| Procedure | Goal | Alternatives | Pitfalls | |
|---|---|---|---|---|
| Receptor Modeling | Correct receptor pocket model(s) | Sources: X-ray, NMR, or homology modeling.Apo-form or liganded-form.Alternative conformations predicted by simulations | Receptor model does not reflect the induced fit. Alternative conformations are missed |
| Library Generation | Sufficiently large and diverse set of relevant compounds | In-house collection, HTS hits, commercially available compounds, virtual libraries computed from accessible scaffolds and side-chains | The library is too restricted, molecules are not chemically feasible or not drug-like |
| Flexible Docking | Correct prediction of the binding geometry | MC or GA, Stochastic global optimization with gradient minimization, Incremental construction, grid or explicit receptor representations, etc. | Inaccurate energy function, poor optimization algorithm. Wrong receptor model, inadequate ligand flexibility. |
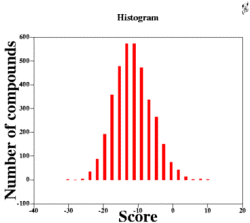
| Ligand Scoring | Maximal separation between binders and non-binders | Weighted interaction terms,Statistical potentials, combination of binding score with QSAR if binders are known | Poorly predicted binding geometries, score over-training to a particular case/family, large number of false positives. |
| Hit List Post-Processing | The best task for the chemist, screener or compound vendor | Clustering, diversity, selection of scaffolds and/or side-chains for a small combinatorial library of parallel synthesis | Domination of one chemical family, lack of chemical availability, or ADME-tox and patent considerations. |
[Curr. Opin. Chemical Biology - 2001] [PDF]