We collaborate with laboratory of Prof. Rik Wierenga from University of Oulu, Finland.
In protein design our goal is to demonstrate that our ICM structure prediction technology can predict new backbone conformations de novo with high accuracy and that the results can be used as guidelines for real in vitro experiments. For example:
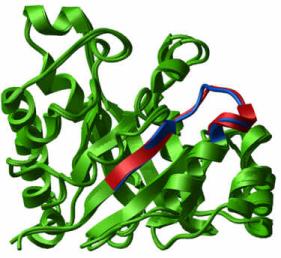
The ICM method was applied to redesign a 15-residue loop-3 of trypanosomal triosephosphate isomerase (TIM) dimer. The purpose of the design was conversion of the dimer into a monomer. Several polypeptide chain fragments of different amino acid sequence and length were tried and the ICM global energy optimization was performed for each of them. An eight-residue connection with sequence GNADALAS replacing the native sequence IAKSGAFTGEVSLPI between positions 68 and 82 was predicted to fold into a strainless loop with an additional helical turn at the N-terminus of helix 3. The modified polypeptide was synthesized and the first suggested variant was experimentally proven to form a stable, monomeric structure with TIM activity [Abs.]. Subsequent crystallographic studies of the redesigned protein, referred to as mono-TIM, demonstrated that the protein retains the characteristic TIM-barrel fold and that the new loop was correctly predicted with a main-chain atom rms difference of 0.4 Ċ for the loop residues [Abs.].
Interestingly, two other loops of the original dimer interface, loop-1 and loop-4, were found to change their conformational state [Abs.]. Loop-1 became disordered, which in turn influenced the ability of an active site residue Lys13 to reach the substrate. This inspired another design project in which loop-1 was rigidified [Abs.]. This loop design was the second blind test of the ICM loop prediction algorithm. A design scheme similar to the scheme used previously for loop-3 was employed. A series of ICM simulations suggested shortening of the eight-residue loop by one residue as well as some modifications of the sequence. The predicted structure was deposited in the PDB and the crystallographic structure was determined. The experimental structure confirmed that the loop became rigid and was predicted correctly. The direct superposition of the lowest energy structure of the proposed loop KSGSPDS to the crystallographic structure results in an rms difference of 0.5 Ċ for the 28 main-chain atoms.
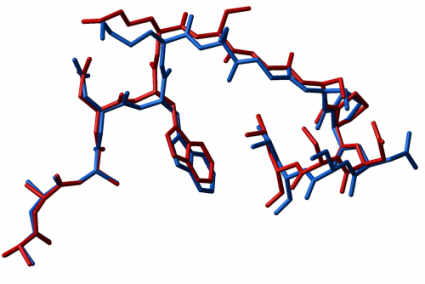 |
| An example of the modelled loop compared to the crystallographically solved structure. |
In summary, two (out of two) blind predictions aimed at designing loop-1 (eight residues) and loop-3 (seven residues) in triosephosphate isomerase were successful examples of the ICM ab initio loop prediction technique. Let us note, however, that these were not single loop predictions, but rather a series of iterative sequence modifications followed by structure predictions. In this setup, even slightly wrong initial predictions can be stabilized by further sequence modifications. Loop predictions in modeling by homology are much more challenging because a sequence cannot be adjusted and, more importantly, because the structural environment (loop ends and surrounding residues) of the loop on a homologous template may be strongly distorted with respect to the true environment [Abs.].