|
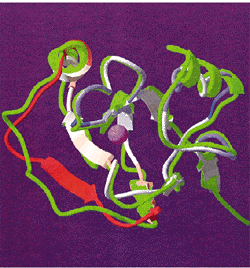
| Correlation of the probability of local structural difference calculated in a window of 11 residues using the analytical formulae for structural significance of sequence alignment with the RMS deviation of the backbone Ca atoms between the template and the experimentally determined structure for T0009. The reliability function is shown by color (blue, reliable; red, unreliable) on a structural superposition of our model of T0009 and the experimental structure (green ribbon). |
Five models by homology containing insertions and deletions and ranging from 33% to 48% sequence identity to the known homologue, and one high sequence identity (85%) model were built for the CASP2 meeting. For all five low identity targets: (i) our starting models were improved by the Internal Coordinate Mechanics (ICM) energy optimization, (ii) the refined models were consistently better than those built with the automatic SWISS-MODEL program, and (iii) the refined models differed by less than 2% from the best model submitted, as judged by the residue contact area difference (CAD) measure [Abagyan, R.A., Totrov, M.J. Mol. Biol. 268:678-685, 1997]. The CAD measure is proposed for ranking models built by homology instead of global root-mean-square deviation, which is frequently dominated by insignificant yet large contributions from incorrectly predicted fragments or side chains. We demonstrate that the precise identification of regions of local backbone deviation is an independent and crucial step in the homology modeling procedure after alignment, since aligned fragments can strongly deviate from the template at various distances from the alignment gap or even in the ungapped parts of the alignment. We show that a local alignment score can be used as an indicator of such local deviation. While four short loops of the meeting targets were predicted by database search, the best loop 1 target T0028, for which the correct database fragment was not found, was predicted by Internal Coordinate Mechanics global energy optimization at 1.2 A accuracy. A classification scheme for errors in homology modeling is proposed.
[Proteins - 1997] [PDF]