|
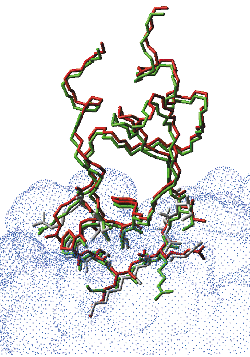
| Docking simulations of the interaction between trypsin and BPTI: 3-D model of the unbound ligand BPTI (backbone and binding side-chains, in green) after rigid-body docking onto the unbound trypsin (receptor surface in blue). The ligand binding side-chains are optimized after the refinement step (shown in red), and can be compared to the conformation of the ligand binding side-chains in the crystallographic structure (in white). |
Association of two biological macromolecules is a fundamental biological phenomenon and an unsolved theoretical problem. In recent years, several groups have developed a variety of tools in an attempt to solve the so-called protein-protein docking problem, that is, the prediction of the geometry of a complex from the atom coordinates of its uncomplexed constituents. These predictive docking methods usually fail when they are applied to a large set of complexes, mostly because of inaccuracies in the scoring function and/or difficulties on simulating the rearrangement of the interacting side-chains of the individual molecules upon binding. Our lab have developed and optimized a two-step docking procedure (pseudo-Brownian rigid body docking [Abs.] followed by Biased Probability Monte Carlo minimization [Abs.] of the ligand interacting side chains) that uses a fast soft interaction energy function pre-calculated on a grid [Abs.]. The use of grid potentials, instead of the explicit energy, increased drastically the speed of the procedure. The method was tested in a benchmark of twenty-four protein-protein complexes where the 3D structures of their subunits (bound and free) are known. The results shown that the rank of the near-native conformation in a list of candidate docking solutions was below 20 in 85 % of complexes with no major backbone motion upon binding. Among them, as many as seven protease-inhibitor complexes out of eleven (64 %) can be successfully predicted as the highest rank conformations.
[Fernandez-Recio, J., Totrov, M. & Abagyan, R., [Protein Sci.]].
A web server of the protein-protein docking algorithm is available here.