Shell Scripts and Awk Programs
cartopdbq
Usage
: cartopdbq lig.car > lig.pdbq
Converts from Biosym InsightII
".car" format to PDBQ format
check-qs
Usage
: check-qs lig
Needs:
lig.pdbq
Creates:
lig.err
Checks partial atomic charges
in PDBQ file; any non-integral charges are reported.
checkqs
Usage
: checkqs lig
Needs:
lig.pdbq
Creates:
lig.err
Sorts the input PDBQ file by
residue number before running the result through
check-qs .
clamp
Usage
: clamp grid.map > grid.map.NEW
Clamps any
AutoGrid
map values that exceed ECLAMP (normally set to 1000.0)
cnvmol2topdbq
Usage
: cnvmol2topdbq lig.mol2 > lig.pdbq
Needs:
lig.mol2
Creates:
lig.pdbq
This converts from (fixed format)
Tripos
SYBYL mol2 fiormat into PDBQ format, but stores all the
residues' chain-IDs specified by the SUBSTRUCTURE records in the mol2 file.
These chain-IDs are then output when the PDBQ lines are written.
deftors
Usage
: deftors lig.mol2
Creates
: lig.pdbq
, lig.err
Sets up rotatable bonds for
AutoDock . This script launches
AutoTors , with the -A +15.0,
-a, -h and -m flags; it also checks the charges in the output PDBQ file,
with
check-qs .
dpf3gen
Usage
: dpf3gen lig.pdbq > lig.dpf
This is normally used by
mkdpf3, so you should not use this script by hand.
It generates a pre-cursor
to a default AutoDockdocking parameter file. You must edit
the file before using it. This reads in the small molecule PDBQ file,
detects all atom types present in the lig.pdbq; and creates a docking parameter
file for AutoDock . Note the user must replace the tags <lig>
and <macromol> by appropriate filename stems.
This uses equilibrium separations
and well depths to define pairwise energy potentials, rather than coefficients.
Usage:
get-coords lig.vol > lig.txt
This is used as part of
prepare, prepare-gpf+dpf, prepare
IIand prepare III. It takes the .volfile
created by pdb-volumeand creates a line that can be used
in the grid parameter file to specify the center of the maps.
get-docked
Usage
: get-docked lig.macro.dlg
Creates
: lig.macro.dlg.pdb
This extracts the docked records
from a docking log file. This is very useful when wanting to view the results
of a docking in a molecular modelling program or molecular viewer. It is
essentially the same as the `
dockedtopdb ' awk program.
gethis
Usage
: gethis lig.macro.dlg
-
This outputs the histogram from the confomational
analysis section of the AutoDock log file, lig.macro.dlg
, and writes it to the screen.
getready
Usage
: getready lig.pdb
Needs:
pdbinfo, pdbsplitchains, pdbwaters, pdbdewater
Creates:
lig.info, lig_.atm.pdb, lig_.het.pdb, lig_.wat.pdb
-
This is a very useful script to get started.
It will split a PDB file into separate files, each containing a different
chain, and will split each of these chains into ATOM, non-water HETATM
and water containing PDB files. Also the .info file is a useful summary
description of the PDB file.
gpf3gen
Usage
: gpf3gen lig.pdbq[s] > lig.gpf
This is used by
mkgpf3
, and should
not be used by hand; otherwise the user must edit certain
tags by hand before this can be used by
AutoGrid .
This generates a precursor to
a grid parameter file. It takes lig.pdbq as its input file, detects
all atom types present, and creates the properly formatted parameter file
for AutoGrid . It uses equilibrium separations and well depths to
define pairwise energy potential. It also assigns atomic solvation parameters,
based on Stouten, P.F.W., FrÖmmel, C., Nakamura, H., and Sander, C.
(1993), "An effective solvation term based on atomic occupancies for use
in protein simulations", Molecular Simulation , 10 , 97-120.
histable
Usage
: histable lig.macro.dlg
Creates
: lig.macro.dlg.tbl
This extracts the histogram
from the docking log file, and counts all the `#' symbols, writing the
result in a table file. This is suitable for input to a variety of graph
drawing programs and spreadsheets.
job3
Usage
: job3 lig.macro > lig.macro.joblog &
Launches a single
AutoDock
3.0 job. It assumes that "
lig.macro.dpf "
exists,
and executes
AutoDock using the arguments:
autodock3 -p lig.macro.dpf -l lig.macro.dlg
You must edit this script
the first time you use it, so that the environment variables $root, $bin
and $sh are correctly set equal to, respectively: the path to the root
of
AutoDock tree, the architecture-dependent binary subdirectory
and the Unix scripts subdirectory. The file
lig.macro.joblog contains
the output from the job script.
makebox
Usage
: makebox macro.gpf >! macro.gpf.box.pdb
Creates
: macro.gpf.box.pdb
-
This creates a PDB file from the grid parameter
file `macro.gpf', that shows how big and where the grid box will be when
AutoGrid calculates the grid maps. You can use this `box molecule' to help
refine the center and number of grid points in the grid maps, before you
run AutoGrid.
If you colour the `box molecule'
by atom type,
i.e. red for oxygen, green for carbon, and blue for
nitrogen, then the edges of this box will be coloured-coded to indicate
the Cartesian axes. R,G,B will correspond to
x,y,z, respectively.
Your molecule viewer must obey the CONECT records in the `
macro.gpf.box.pdb
' file, even though the corresponding bonds may appear too long, otherwise
the edges of the grid box will not be displayed.
mkbox
Usage
: mkbox macro.gpf >! macro.gpf.box.pdb
Creates
: macro.gpf.box.pdb
This is very similar to `
makebox ', except that this puts a
phosphorus atom at the minimum
x , minimum
y and minimum
z coordinates of the box.
This helps to convey which directions are
+x, +y and
+z .
Once again, if oxygen is red, carbon is green and nitrogen is blue, then
R,G,B will correspond to
x,y,z, respectively.
mkdlgfld
Usage
: mkdlgfld lig.macro.dlg
Needs
: lig.macro.dlg
Creates
: lig.macro.dlg.fld
Only needed for AVS users.
This extracts the "AVSFLD" records
from an AutoDock log file, and puts them in lig.macro.dlg.fld. These
"AVSFLD" descriptors must be removed before the file can be used in AVS.
mkdpf3
Usage
: mkdpf3 lig.pdbq macromol.pdbqs
Needs
: dpf3gen, dpf3gen.awk (AWK program)
Creates
: lig.macro.dpf
This creates a default docking
parameter file for
AutoDock 3.0; it needs the ligand in PDBQ format
and the macromolecule in PDBQS format. It uses the script
dpf3gen
, which in turn calls the
awk program `
dpf3gen.awk '.
The
lig.macro.dpf docking parameter file is based on the atom
types detected in the input
lig.pdbq file. See
dpf3gen
above.
mkgpf3
Usage
: mkgpf3 lig.pdbq macromol.pdbqs
Needs
: gpf3gen, gpf3gen.awk (AWK program),
pdbcen (AWK program)
Creates
: macro.gpf
This creates a default grid
parameter file for
AutoGrid 3.0; it needs
gpf3gen.awk and
pdbcen , both
awk programs. See
gpf3gen above.
mol2fftopdbq
Usage
: mol2fftopdbq lig.mol2 > lig.pdbq
Needs:
lig.mol2
Creates:
lig.pdbq
Converts from free formatted
SYBYL mol2 into
AutoDock PDBQ format. Chain-IDs specified in the
mol2 file by the
SUBSTRUCTURE records are incorporated into the
PDBQ file.
mol2topdbq
Usage
: mol2topdbq lig.mol2
Needs:
lig.mol2
Creates:
lig.pdbq
Converts from fixed-format
SYBYL mol2 into
AutoDock PDBQ format, and automatically names the
output based on the stem of the input mol2 file. Do not use "
mol2topdbq
lig.mol2 > lig.pdbq ", because "
lig.pdbq " is automatically
created.
mol2topdbqs
Usage
: mol2topdbqs lig.mol2
Needs:
lig.mol2
Creates:
lig.pdbqs
Converts from SYBYL mol2 format into AutoGrid
3.0 PDBQS format, by calling mol2topdbq then running addsol
on the intermediate PDBQ file. Like mol2topdbq , it also removes
any lone pairs (using " rem-lp "), and automatically names the
output based on the stem of the input mol2 file. There is no need to use
" mol2topdbq lig.mol2 > lig.pdbq ", because " lig.pdbqs
" is automatically created.
pdbcen
Usage
: pdbcen lig.pdb
Creates
: a "gridcenter" line in AutoGrid GPF format, holding the x,y,z coordinates of the molecule.
This calculates the center
of a molecule supplied in PDB format, and outputs a line holding the x,y,z
coordinates of the molecule for inclusion in an
AutoGrid 3.0 grid
parameter file (GPF).
pdb-center
Usage
: pdb-center [ lig.pdb | lig.pdbq ] > lig2.pdb
Calculates the center of mass
of each residue; writes these coordinates out using REMARK records.
pdb-center-all
Usage
: pdb-center-all [ lig.pdb | lig.pdbq ] > lig2.pdb
Calculates the center of mass
of each residue; writes these coordinates out using REMARK records.Also
calculates the center of all the residues.
pdb-distance
Usage
: pdb-distance macro.pdb
The first line of the
macro.pdb
file defines the center of the distance profile. It is just a copy of the
line containing the atom of interest, which will be the origin for the
distance calculations. However, it must have the ATOM or HETATM record
replaced with a non-PDB tag, `FROM'. The x,y,z coordinates in this FROM
line will then be used to calculate the distance to the center of each
residue in the protein. Finally, this awk program outputs a bar chart using
`#' symbols, showing the distance from this point to each residue. This
can be useful to identify all the residues nearest a particular ligand
atom, or near an active site.
pdbdewater
Usage
: pdbdewater macro.pdb >! macro.dry.pdb
This removes any water records
from a PDB file.
pdbinfo
Usage
: pdbinfo macro.pdb
Builds a summary of the contents
of a PDB file.
pdb-volume
Usage
: pdb-volume [ lig.pdb | lig.pdbq ] > lig2.pdb
Calculates the center of mass
of each residue. Writes out REMARKs showing these coordinates. Draws ASCII
diagram showing volume extents of each residue.
pdbqtobnd
Needs:lig.pdbq
Creates:lig.bnd
Creates " lig.bnd " from
the existing " lig.pdbq " ligand PDBQ file. Note this script needs
just the stem of the file name. This script executes " pdbqtoatm
" and " atmtobnd ": the latter is an executable, not a script,
so it must be compiled for each architecture and operating system used.
pdbqtopdb
Usage
: pdbqtopdb lig.pdbq > lig.pdb
Converts from
AutoDock
PDBQ to PDB format.
pdbsplitchains
Usage
: pdbsplitchains macro.pdb
Creates separate PDB files
that contain each of the chains in
macro.pdb . The chain IDs are
used to name the new PDB files. If there is no chain ID, the underscore
character, `_' is used.
pdbtoatm
Usage :
pdbtoatm
lig.pdbq > lig.atm
This creates a Connolly ATM formatted
file " lig.atm " from the ligand PDBQ file, " lig.pdbq
". This is used to create input for the utility program atmtobnd
to generate a bond connectivity file.
pdbdewaters
Usage
: pdbwaters macro.pdb > macro.wet.pdb
-
Extracts the waters from macro.pdb into
macro.wet.pdb .
prepare
Usage:
prepare m s
where: m.pdb and
s.pdbq contain the receptor and ligand respectively.
Prepare
performs the following eight steps. The macromolecule
`.pdb '
filename stem is represented by "
m ", and the ligand
`.pdbq
' filename stem by "s":
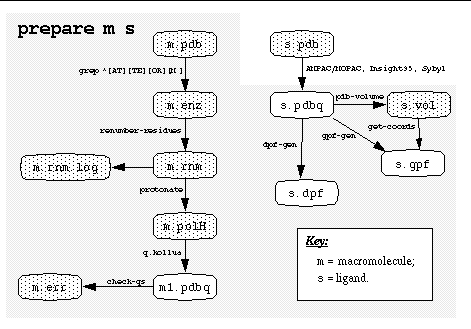
1. Extracts all ATOM and TER
records from
m.pdb into
m.enz;
2. Renumbers residues to avoid problems in protonate -step;
3. Adds polar hydrogens to m.enz , creating m.polH
;
4. Somewhat crudely assigns partial atomic charges to m.polH
, creating m.pdbq ;
5. Checks charges in m.pdbq , all errors held in m.err;
6. Creates s.gpf , a parameter file for AutoGrid
, based on ligand file s.pdbq ;
7. Creates s.vol , a volume dimensions file; and finally,
8. Creates s.dpf , a parameter file for AutoDock, based
on ligand file s.pdbq ;
Its arguments are the stem of
the filename of the macromolecule ` .pdb ' file and that of the
ligand PDBQ file. See the flowchart below for more details. It shows what
files are created by ` prepare ', and which scripts or programs
are used. Steps 1.-4. are better carried out with a reliable molecular
modeling system: these steps can produce some odd results unless carefully
checked.
The user must check the
m.err error file to ensure there are no non-integral charges, either
on any residue in the macromolecule, or on the macromolecule as a whole.
If there are, then the user must repair the m.pdbq file. This
problem can arise if there are atoms for which no coordinates were assigned
by the crystallographer, e.g . due to ambiguous electron density.
Assuming there were no problems, s.gpf and s.dpf should
be successfully produced.
prepare-gpf+dpf
Usage
: prepare-gpf+dpf macro lig
Executes only steps 6. through
8.
rem-lp
Usage
: rem-lp lig.pdbq
or
: rem-lp lig.mol2
Creates:
lig.pdbq
or
: lig.mol2
This removes the lone-pairs
(atom name = LP) added by some molecular modelling programs, such as SYBYL,
and adds their partial charges on to that of the atom to which they were
attached (SG in cysteines and SD in methionines). Otherwise, AutoDock treats
lone-pairs as carbon atoms. (Note: if you need lone-pairs, you can force
AutoGrid to calculate a grid map for "LP" atoms, using the atom code "L"
in the "types" commands of AutoGrid and AutoDock).
renumberatoms
Usage
: lig.pdb > lig2.pdb
-
Used to renumber the atom IDs in the first column
of the ATOM and HETATM records of a PDB file. Also updates the CONECT records
appropriately.
-
renumber-residues
Usage
: lig.pdb > lig.rnm
Used by
prepare to
renumber residues in the macromolecule contiguously. This step is needed
prior to using
protonate , which may fail if there are gaps in
the residue numbers.
resrange
Usage
: resrange lig.pdb
This is handy to summarise
the range(s) of residues in a given protein PDB file.
runtrj
Usage
: runtrj lig
Needs:
lig.dpf
, lig.trj
Creates:
lig.tcom
, lig.tlg
and
lig.tout
This creates an
AutoDock
command file,
lig.tcom , which is then used to convert the trajectory
written in state variables (
lig.trj ), into a trajectory written
in cartesian coordinates.
lig.trj is created by an earlier run
of
AutoDock , in which
trjfrq was set to a non-zero value.
stats
Usage
: stats columns.dat
This is a very useful, general
awk program. Use it to calculate the minimum, maximum, mean and
standard deviation for each column of numbers in an input file, here `
columns.dat '. Any alphanumeric columns will be ignored.